

Plant PTM Viewer Tutorials
Protein Search
DownloadUsers have three options to find a protein of interest: (a) search by protein identifier, (b) search by sequence (here ubiquitin), or (c) search by protein description. In addition, the search can be restricted to a single plant species by using the dropdown menu selection option below.
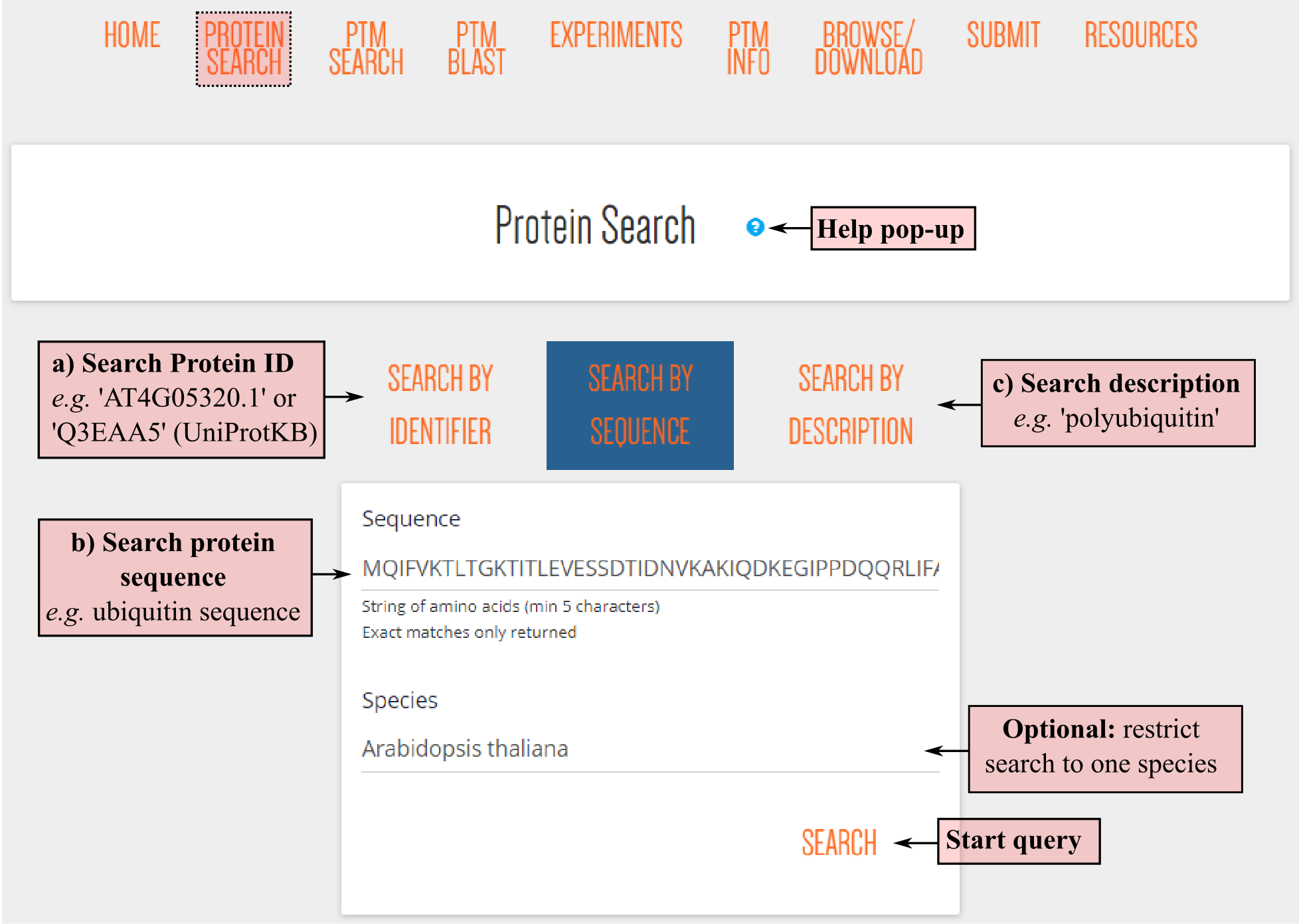
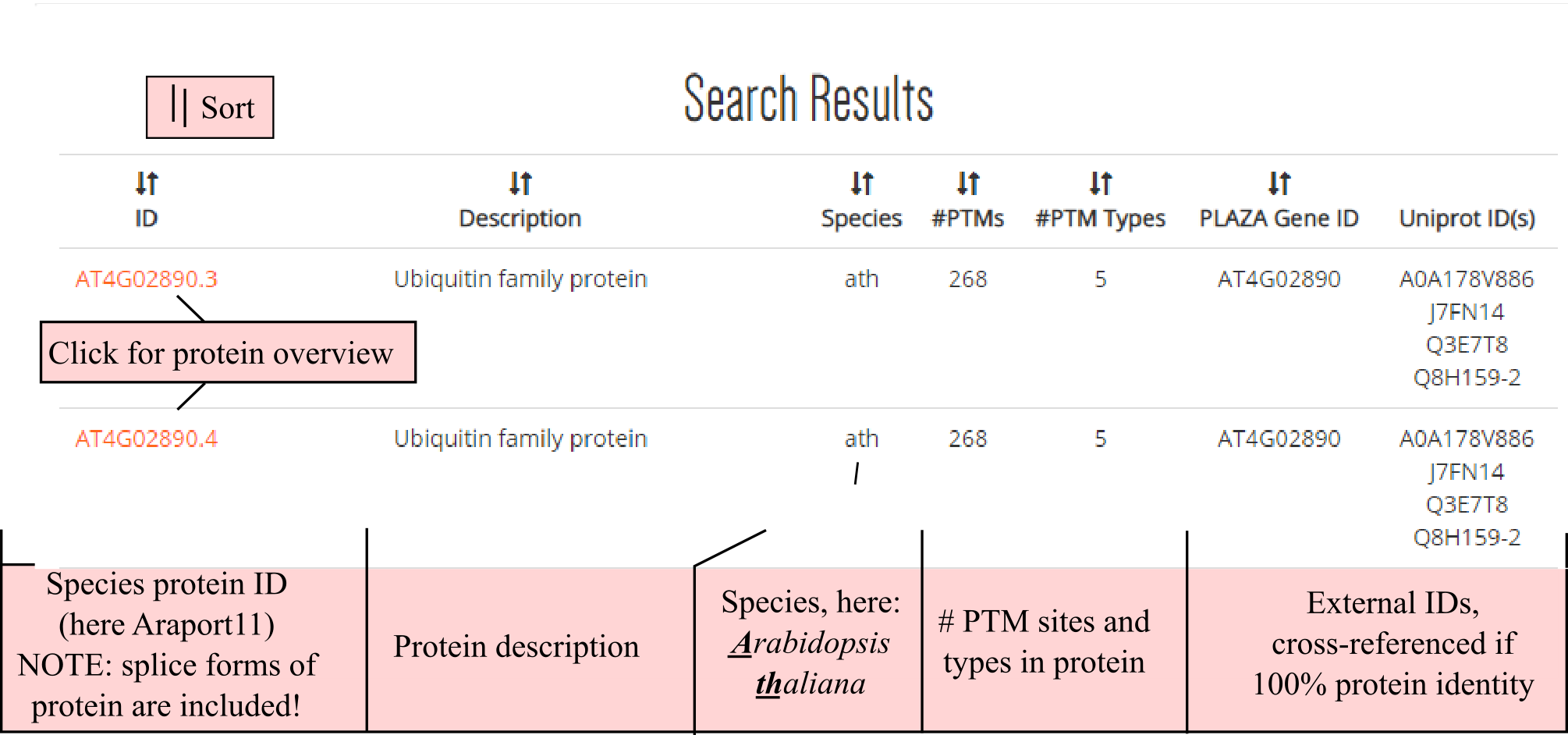
After pressing the SEARCH button, any results will appear below the query box. All proteins fulfilling the criteria will be listed in the search results table. Note that the result table also includes protein splice forms! All columns can be sorted, including a description column, species abbreviation, cross-references protein identifiers of PLAZA or UniProtKB (requiring identical protein sequence) or the amount of PTM sites and types.
By clicking a protein identifier the PTM protein sequence overview is launched, as example we show here the protein encoded by polyubiquitin 10 (AT4G05320.1). Below a general protein info header with description and cross-references, a PTM table (left, green border), PTM protein sequence overview (top-right, red border), and protein domain/site table is provided (bottom-right, blue border).
These tables are interactively connected to each other. For instance, by default all PTM checkboxes are selected in the PTM table. Removing a specific checkbox will remove the highlighting in the protein sequence overview. Note that a color legend can be displayed and also by hovering over a modified amino acid, the modification(s) will appear in a pop-up box. Similarly, a protein domain can be selected, e.g. here all ubiquitin domains were selected, and the domain will be underlined in the PTM protein sequence overview.
In the PTM table additional information is found, such as the type of PTM with corresponding protein position, the originating (plain) peptide identified by MS, the respective publication and a confidence color-coding. By clicking the MS study, the experiment overview is launched. If localization probabilities or differential abundance estimates (log2 fold change and significance) are available, these are displayed as well. Log2 fold changes are displayed in a heatmap-like gradient (green is upregulated, red is downregulated). In case the significance estimate was below the threshold employed in the respective study, this is also highlighted in green (note this was not the case here).
The PTM table can be exported by clicking the EXPORT RESULTS button.

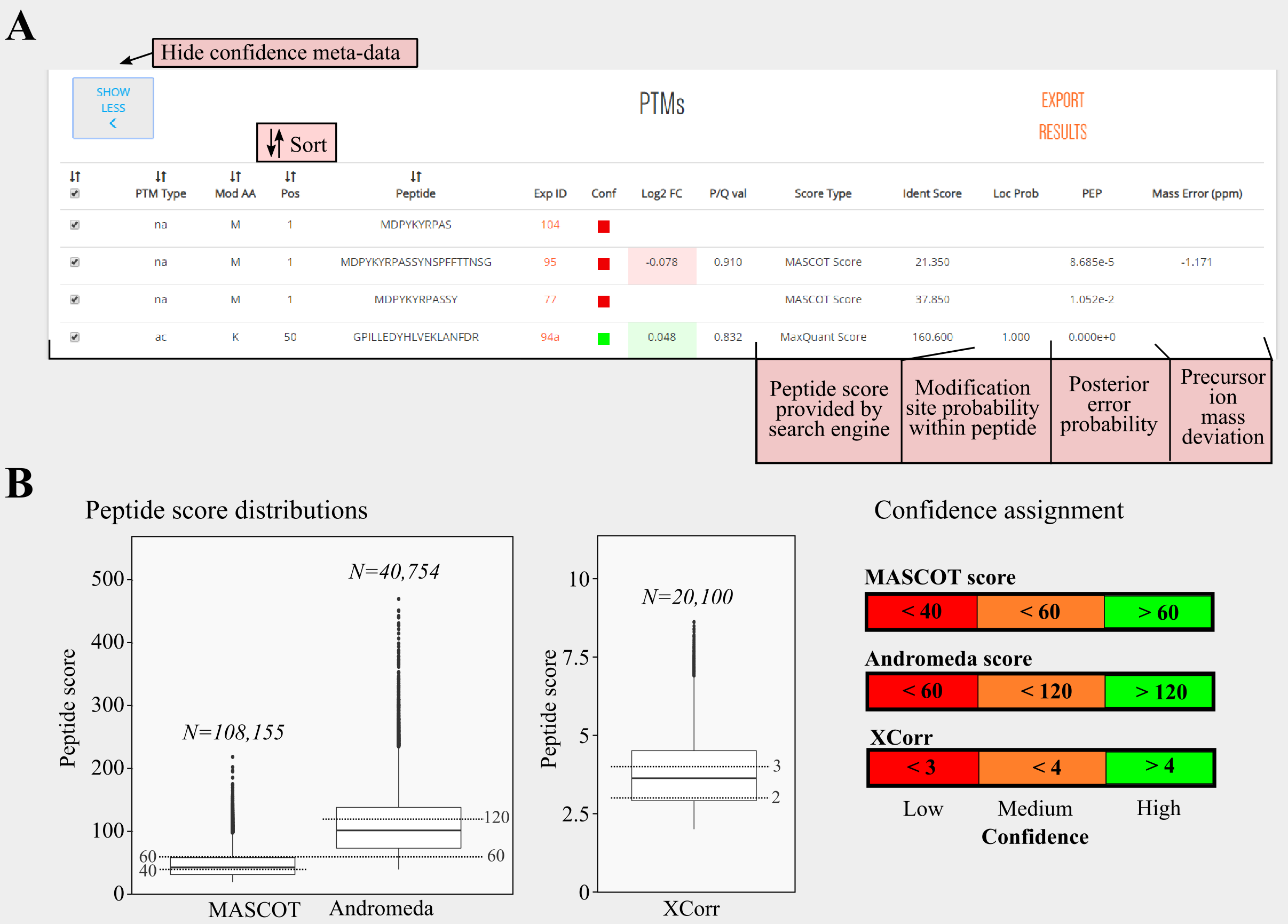
Details of the confidence meta-data collected can be consulted by clicking SHOW CONFIDENCE. Below, we can view the extended version (figure panel A) including these confidence estimates reported by experiments, including peptide scores, posterior error probability (PEP), modification site localization probability and/or precursor mass deviation.
Peptide scores are measured by search engines and score how a tandem mass spectrum matches a peptide from the searched protein database. Most frequently reported scores (used search algorithms) are the MASCOT ion score (MASCOT, Perkins et al., 1999), the Andromeda score (built-in MaxQuant software suite, Cox et al. 2011) and the cross-correlation score (XCorr, originally for SEQUEST, Eng et al., 1994). Distributions of these scores can be consulted in the figure panel B below. For these three search engines minimal peptide score thresholds were used. MASCOT ion scores were required to be at least 20, Andromeda scores 40 and XCorr scores at least 2.
Next to peptide scores, which are highly differing and dependent on the search algorithm used, the PEP provides a more unified confidence estimate and can be considered as a "local FDR" that expresses the chance that a given peptide-to-spectrum match was incorrect. Most PEP values reported here were measured by software such as MaxQuant (Cox and Mann 2008), Proteome Discoverer (Thermo Scientific) or post-processing algorithms such as Percolator (Käll et al., 2007).
Lastly, beside peptide-level confidence measurements, modification localization probability within a peptide can be assessed by algorithms such as PhosphoRS (Taus et al., 2011) or the PTM Score implemented in MaxQuant (Olsen et al., 2006). Here, we required a modification site localization probability of at least 0.75, when reported. Based on the peptide scores provided, PTMs are categorized as being low, medium or high confident (figure panel B - right). Assessing reliability of PTMs is a crucial step as false positive identification may occur in mass spectrometry identification results. In this aspect, careful inspection of experimental details remains therefor advisable.
REFERENCES
- Cox, J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V. and Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794-1805.
- Eng, J.K., McCormack, A.L. and Yates J.R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976-989.
- Käll, L., Canterbury J.D., Weston J., Noble W.S. and MacCoss M.J. (2007). Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923-925.
- Olsen, J.V., Blagoev, B., Gnad, F., Macek, B., Kumar, C., Mortensen, P. and Mann, M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635-648.
- Perkins, D.N., Pappin, D.J., Creasy, D.M. and Cottrell, J.S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551-3567.
- Taus, T., Köcher, T., Pichler, P., Paschke, C., Schmidt, A., Henrich, C. and Mechtler, K. (2011) Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354-5362.
PTM Search
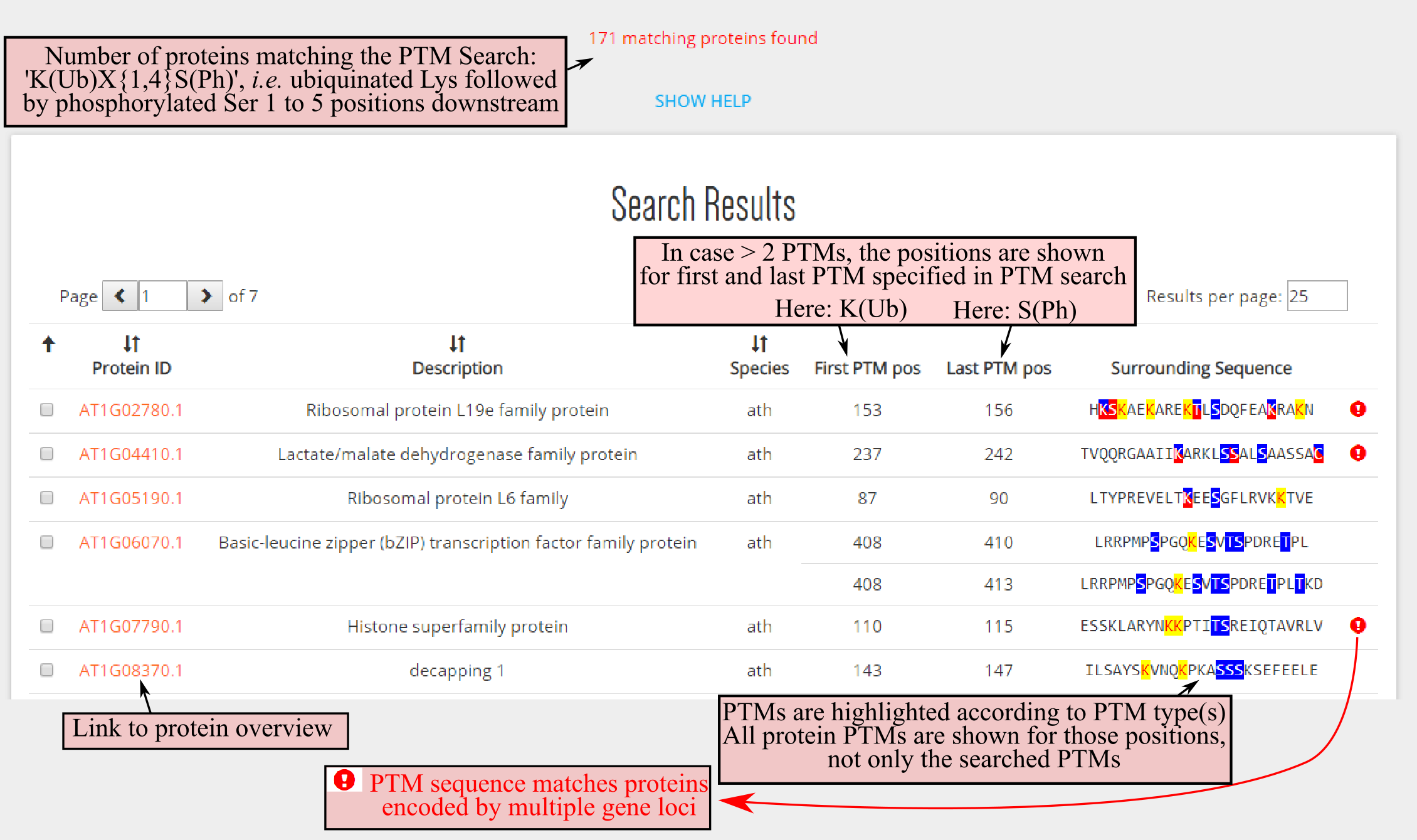
DownloadPTM search allows users to retrieve PTMs in a specific amino acid sequence context and/or combinations of PTMs. This is possible thanks to a regular expression search that allows to search for ambiguity in PTM type and amino acids. As example we consider the search K(Ub)X{1,4}S(Ph), i.e. a ubiquinated lysine, K(Ub), followed maximal five amino acids downstream, X{1,4}, by a phosphorylated serine S(Ph).For more details regarding the regular expression please consult the extensive help section below on the PTM Search page.
Besides inputting the PTM Search query, several advanced options can be fine-tuned. For instance, resulting PTMs can be required to be resorting from peptides matching proteins encoded from a single gene, restrict the search to a single plant species and the eventually displayed sequence window length.
In addition, there are advanced options that require PTMs to reside in specific protein regions. For instance, PTMs can be required to reside in a specific InterPro protein domain, by inputting the InterPro identifier, or match a UniProtKB site-specific annotation. For the site annotations, a selection to four types: active site, binding site, metal ion-binding site and site. For more info regarding these site annotations we refer to the UniProtKB help section (https://www.uniprot.org/help/sequence_annotation).
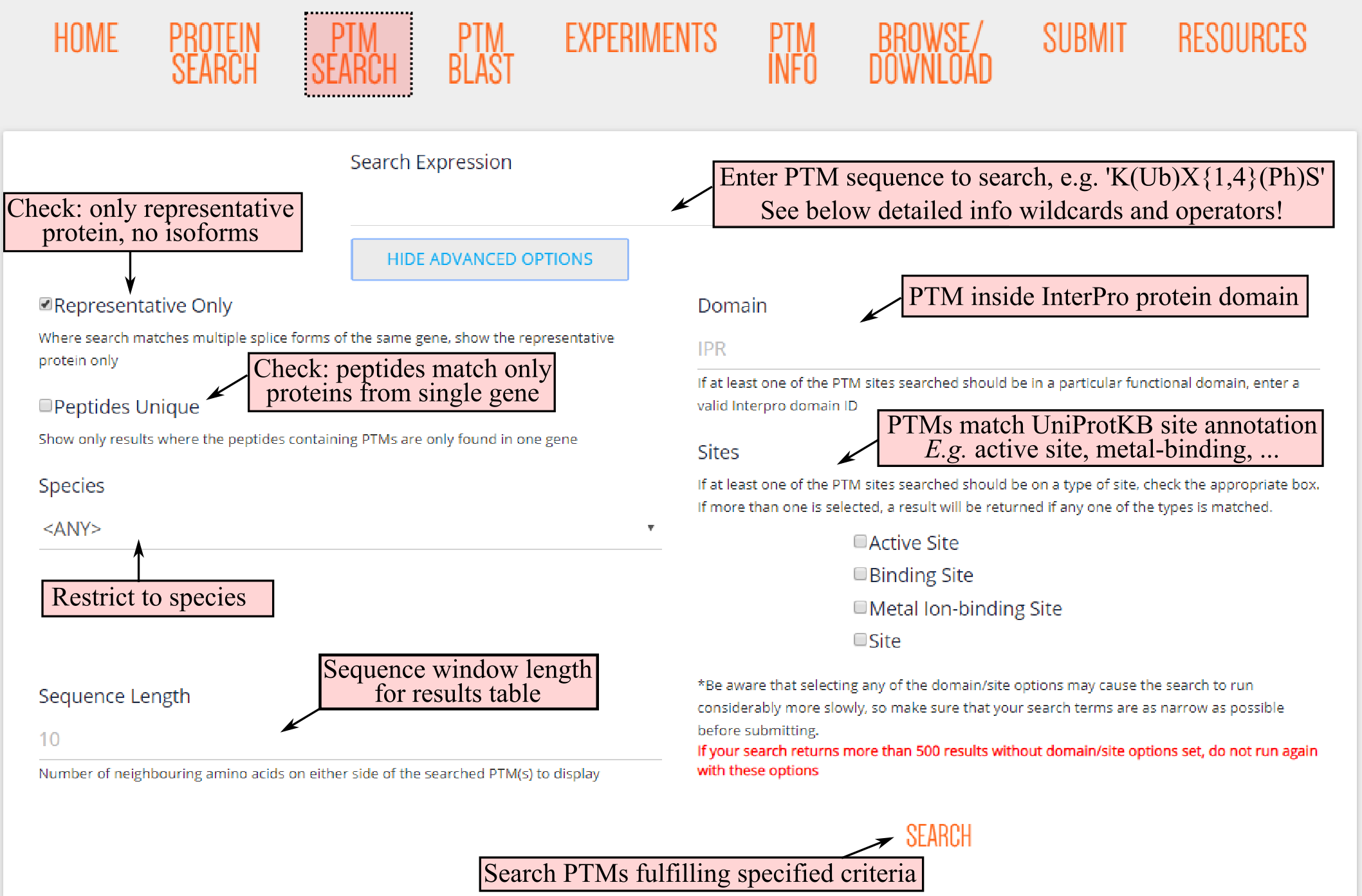
After clicking the SEARCH button, PTM Search will be executed and retrieve matching PTM sequences. Note that for highly ambiguous searches, e.g. many (xx) as PTMs or X as amino acid, the PTM search might take some time. This is especially true for PTM searches restricted to protein domains or UniProtKB annotated sites. We are currently optimizing the speed for these last search options.
After the PTM Search is finished, a search result table will appear on the bottom of the page. This table is organized across multiple pages and the results per page can be customized. The results are organized per protein and above the total amount of proteins that contain the searched PTM sequence are displayed. Some proteins might have multiple PTM sequences fulfilling the rules. For instance, for our search, AT1G06070.1 a phosphoserine is found 2 and 5 positions after a ubiquinated lysine.
The respective PTM positions within the protein are displayed for the first and last PTM specified in the PTM search. In case a single PTM was searched, logically only one PTM position is returned. In the returned PTM sequence window, which length can be customized (see above), all PTMs are highlighted according PTM type. In case if the PTM sequence matches proteins encoded from multiple gene loci, these will be flagged. Protein identifiers in the left column provide a link to the respective protein overview page.
PTM BLAST
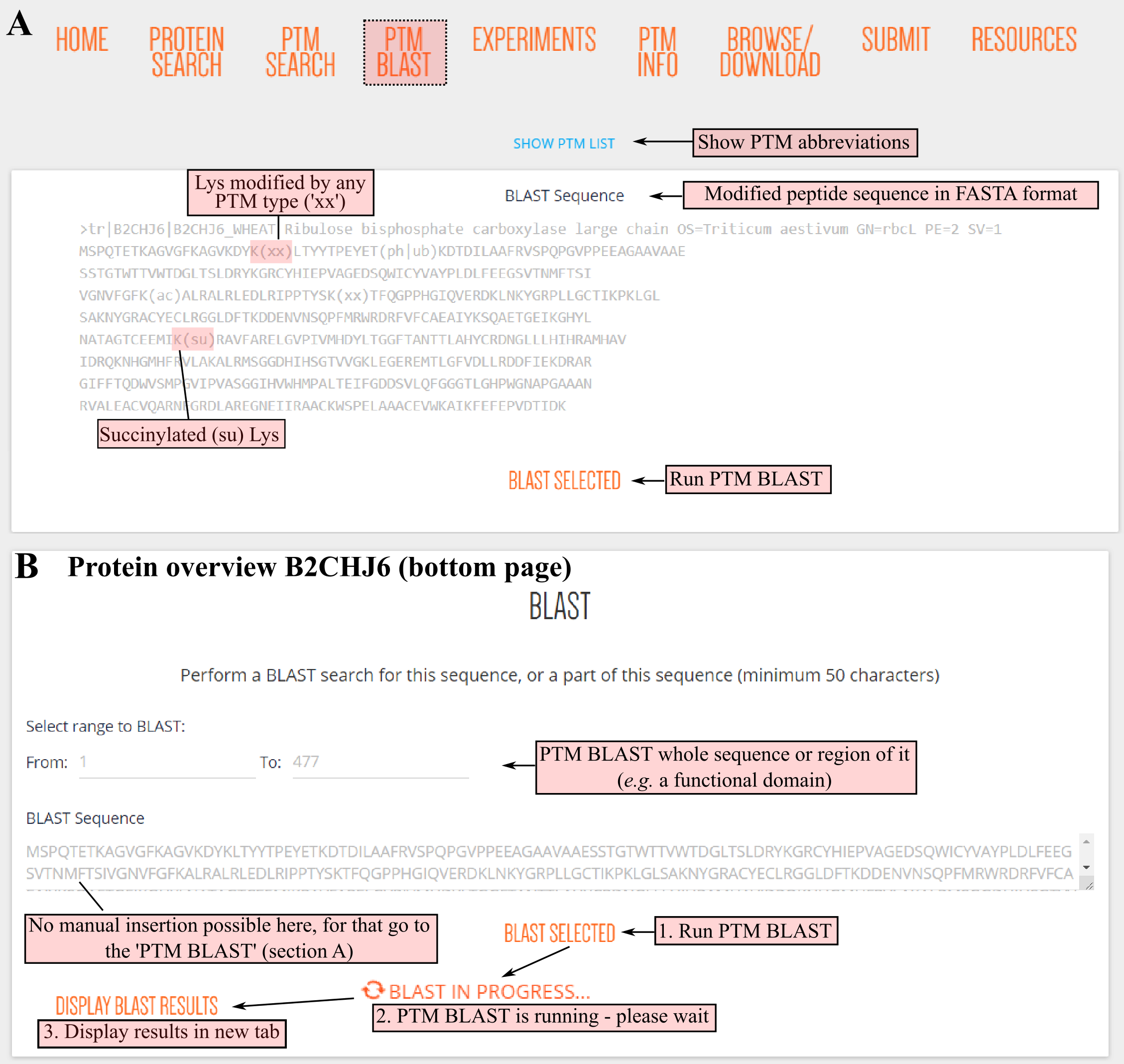
DownloadPTM BLAST allows users to retrieve and compare PTMs in aligned protein sequences. There are two options to initiate PTM BLAST, either from (a) the PTM BLAST section or (b) any PTM Viewer plant protein overview page.
In case of the PTM BLAST section (a), users can specify a protein sequence and indicate PTMs by their two-letter code abbreviation between brackets after the modified amino acid. For instance, phosphothreonine is indicated as T(ph). Any PTM modification can be indicated as T(xx), or multiple can be indicated as T(ph|og).
PTM BLAST can also be launched from any PTM Viewer protein overview (b). Here, the search is restricted to the protein sequence and its PTMs. However, a region from the protein sequence can be searched by indicating the start and end protein positions. This can be useful to PTM BLAST a protein region of interest, for instance a protein domain.
After clicking BLAST SELECTED, a default protein BLAST (BLASTP) will be performed of the plain protein sequence. A progress message will appear and when the BLASTP job is finished the results can be viewed in a new tab by clicking DISPLAY BLAST RESULTS.
The PTM BLAST results are organized in two interactive boxes. In the top PTM BLAST result table, signficant BLASTP hits are displayed and sorted according to number of aligned PTMs. A distinction is made between aligned PTMs, which are aligned amino acid residues modified by the same or different PTM types, and PTM matches, where amino acids are aligned and modified by the same PTM type.
On the left-hand side of the table, PTM BLAST hits can be selected. By default the top three PTM BLAST results, i.e. those with the most PTM alignments, are displayed.
Alignment of proteins and their PTMs are found in the bottom sequence alignment box. Here, for each PTM BLAST hit the BLASTP protein alignment is given and protein sequences are decorated with their PTMs. This display is similar to the protein overview, where PTMs are highlighted in color according their PTM type(s). A color legend can be found below of the page.
In case PTM type(s) matched at a certain aligned amino acid residue, the middle row will also be highlighted to indicate a PTM match. Note that above in the sequence alignment box there is an option to solely display peptides matching protein products from a single gene. In case of a PTM BLAST alignment between plant proteins from the same species, this can filter out PTMs derived from peptides that match both proteins ambiguously.
Both the PTM BLAST results and sequence alignment box are scrollable, thus to view more selected alignments make sure to scroll down in the sequence alignment box.
